【前注】这篇文章有歧义,翻译过来看看。原文是Google翻译,除了一段有歧义用红色纠正,其余照搬谷歌翻译。
研究通过基因突变绘制COVID-19的"早期超新星"图谱,因为它从中国和亚洲传播到澳大利亚、欧洲和北美。研究人员说,他们的方法可以用来帮助识别无证感染源。
来自英国剑桥和德国的研究人员利用基因网络技术,重建了人类COVID-19的早期"进化路径"——感染从武汉蔓延到欧洲和北美。
通过分析首批160个完整的病毒基因组,从人类患者序列,科学家们已经绘制了一些新的冠状病毒的原始传播通过其突变,创造了不同的病毒谱系。
"有太多的快速突变,无法整齐地追踪COVID-19家谱。我们使用数学网络算法同时可视化所有看似合理的树木,"剑桥大学的遗传学家彼得·福斯特博士说。
"这些技术大多以绘制史前人类种群的运动图谱而闻名,通过DNA。我们认为,这是首次使用它们追踪冠状病毒(如COVID-19)的感染途径。
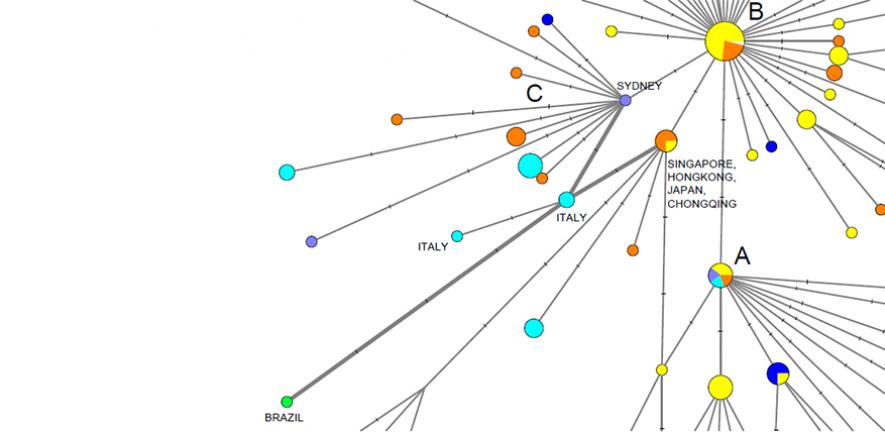
该团队使用2019年12月24日至2020年3月4日期间从世界各地取样的病毒基因组数据。研究揭示了COVID-19的三种截然不同的"变种",包括紧密相关的谱系集群,它们将其标记为"A"、"B"和"C"。
福斯特和他的同事发现,最接近在蝙蝠中发现的COVID-19类型——"A型","原始人类基因组"——存在于武汉,但令人惊讶的是,它不是这个城市的主要病毒类型。
据报道,在武汉居住的美国人中,发现了"A"的突变版本,在美国和澳大利亚的患者中发现了大量A型病毒。
武汉的主要病毒类型"B"在东亚各地患者中流行。然而,研究人员说,没有进一步突变,这种变异没有游出该地区多少,这意味着武汉会发生"创始人事件",或者东亚以外地区对此类COVID-19进行了"抵抗"。
"C"变种是欧洲的主要类型,在法国、意大利、瑞典和英国的早期患者中发现。该研究在中国大陆的样本中不存在,但在新加坡、香港和韩国都能看到。
新的分析还表明,最早将病毒引入意大利之一是1月27日德国首次感染病毒,另一条早期意大利感染途径与"新加坡集群"有关。
重要的是,研究人员说,他们的基因网络技术准确地追踪了既定的感染途径:突变和病毒谱系连接了已知病例之间的点。
因此,科学家们认为,这些"遗传"方法可以应用于最新的冠状病毒基因组测序,以帮助预测未来全球疾病传播和激增的热点。
剑桥麦克唐纳考古研究所研究员福斯特和大学继续教育研究所研究员福斯特说:"遗传网络分析有可能帮助识别无证COVID-19感染源,然后可以隔离,以遏制该病在全球的进一步传播。
研究结果今天发表在《国家科学院学报》(PNAS)上。该研究中使用的软件,以及超过1000个冠状病毒基因组和计数的分类,在www.fluxus-technology.com免费提供。
变异"A"与蝙蝠和穿山甲中发现的病毒关系最为密切,被研究人员描述为"爆发的根源"。类型"B"派生自"A",由两个突变分离,然后"C"依次是"B"的"子"。
研究人员说,"B"变种对东亚的本地化可能是由"创始人效应"引起的:当病毒从一小群孤立的感染中建立一种新类型时,就会发生一种基因瓶颈。
福斯特认为,还有另一种解释值得考虑。"武汉B型病毒在免疫学或环境上可以适应东亚大部分人口。它可能需要变异,以克服东亚以外的阻力。在这个初始阶段,我们看到东亚的突变率比其他地方要慢。
他补充说:"我们详述的病毒网络是流行病早期阶段的快照,在COVID-19的进化路径被大量突变所掩盖之前。这就像在行动中捕捉一个初的超新星。
自今天进行PNAS研究以来,研究小组将其分析扩展到1,001个病毒基因组。福斯特说,虽然还有待同行审查,但最新研究表明,COVID-19的首次感染和传播发生在9月中旬至12月初之间。
研究人员使用的植物遗传网络方法——允许在一个简单的图形中同时可视化数百种进化树——于1979年在新西兰率先出现,然后由德国数学家在20世纪90年代开发。
1998年,考古学家科林·伦弗鲁教授注意到了这些技术,他是PNAS新研究的合著者。伦弗鲁接着在剑桥大学建立了世界上第一个古遗传学研究小组。
《COVID-19: genetic network analysis provides ‘snapshot’ of pandemic origins》
研究通过基因突变绘制COVID-19的"早期超新星"图谱,因为它从中国和亚洲传播到澳大利亚、欧洲和北美。研究人员说,他们的方法可以用来帮助识别无证感染源。
Study charts the “incipient supernova” of COVID-19
through genetic mutations as it spread from China and Asia to Australia,
Europe and North America. Researchers say their methods could be used
to help identify undocumented infection sources.
来自英国剑桥和德国的研究人员利用基因网络技术,重建了人类COVID-19的早期"进化路径"——感染从武汉蔓延到欧洲和北美。
Researchers from Cambridge, UK, and Germany have reconstructed the
early “evolutionary paths” of COVID-19 in humans – as infection spread
from Wuhan out to Europe and North America – using genetic network
techniques.
通过分析首批160个完整的病毒基因组,从人类患者序列,科学家们已经绘制了一些新的冠状病毒的原始传播通过其突变,创造了不同的病毒谱系。
By analysing the first 160 complete virus genomes to be sequenced
from human patients, the scientists have mapped some of the original
spread of the new coronavirus through its mutations, which creates
different viral lineages.
"有太多的快速突变,无法整齐地追踪COVID-19家谱。我们使用数学网络算法同时可视化所有看似合理的树木,"剑桥大学的遗传学家彼得·福斯特博士说。
“There are too many rapid mutations to neatly trace a COVID-19 family
tree. We used a mathematical network algorithm to visualise all the
plausible trees simultaneously,” said geneticist Dr Peter Forster, lead
author from the University of Cambridge.
"这些技术大多以绘制史前人类种群的运动图谱而闻名,通过DNA。我们认为,这是首次使用它们追踪冠状病毒(如COVID-19)的感染途径。
“These techniques are mostly known for mapping the movements of
prehistoric human populations through DNA. We think this is one of the
first times they have been used to trace the infection routes of a
coronavirus like COVID-19.”
该团队使用2019年12月24日至2020年3月4日期间从世界各地取样的病毒基因组数据。研究揭示了COVID-19的三种截然不同的"变种",包括紧密相关的谱系集群,它们将其标记为"A"、"B"和"C"。The team used data from virus genomes sampled from across the world
between 24 December 2019 and 4 March 2020. The research revealed three
distinct “variants” of COVID-19, consisting of clusters of closely
related lineages, which they label ‘A’, ‘B’ and ‘C’.
福斯特和他的同事发现,最接近在蝙蝠中发现的COVID-19类型——"A型","原始人类基因组"——存在于武汉,但令人惊讶的是,它不是这个城市的主要病毒类型。
Forster and colleagues found that the closest type of COVID-19 to the
one discovered in bats – type ‘A’, the “original human virus genome” –
was present in Wuhan, but surprisingly was not the city’s predominant
virus type.
据报道,在武汉居住的美国人中,发现了"A"的突变版本,在美国和澳大利亚的患者中发现了大量A型病毒。
Mutated versions of ‘A’ were seen in Americans reported to have lived
in Wuhan, and a large number of A-type viruses were found in patients
from the US and Australia.
武汉的主要病毒类型"B"在东亚各地患者中流行。然而,研究人员说,这种变异没有进一步突变就游出该地区(没有进一步突变,这种变异没有游出该地区多少),这意味着武汉会发生"创始人事件",或者对东亚以外的此类COVID-19进行"抵抗"(或者东亚以外地区对此类COVID-19进行了"抵抗")。
Wuhan’s major virus type, ‘B’, was prevalent in patients from across
East Asia. However, the variant didn’t travel much beyond the region
without further mutations – implying a "founder event" in Wuhan, or
“resistance” against this type of COVID-19 outside East Asia, say
researchers.
"C"变种是欧洲的主要类型,在法国、意大利、瑞典和英国的早期患者中发现。该研究在中国大陆的样本中不存在,但在新加坡、香港和韩国都能看到。
The ‘C’ variant is the major European type, found in early patients
from France, Italy, Sweden and England. It is absent from the study’s
Chinese mainland sample, but seen in Singapore, Hong Kong and South
Korea.
新的分析还表明,最早将病毒引入意大利之一是1月27日德国首次感染病毒,另一条早期意大利感染途径与"新加坡集群"有关。
The new analysis also suggests that one of the earliest introductions
of the virus into Italy came via the first documented German infection
on January 27, and that another early Italian infection route was
related to a “Singapore cluster”.
重要的是,研究人员说,他们的基因网络技术准确地追踪了既定的感染途径:突变和病毒谱系连接了已知病例之间的点。
Importantly, the researchers say that their genetic networking
techniques accurately traced established infection routes: the mutations
and viral lineages joined the dots between known cases.
因此,科学家们认为,这些"遗传"方法可以应用于最新的冠状病毒基因组测序,以帮助预测未来全球疾病传播和激增的热点。
As such, the scientists argue that these “phylogenetic” methods could
be applied to the very latest coronavirus genome sequencing to help
predict future global hot spots of disease transmission and surge.
剑桥麦克唐纳考古研究所研究员福斯特和大学继续教育研究所研究员福斯特说:"遗传网络分析有可能帮助识别无证COVID-19感染源,然后可以隔离,以遏制该病在全球的进一步传播。
“Phylogenetic
network analysis has the potential to help identify undocumented
COVID-19 infection sources, which can then be quarantined to contain
further spread of the disease worldwide,” said Forster, a fellow of the
McDonald Institute of Archaeological Research at Cambridge, as well as
the University’s Institute of Continuing Education.
研究结果今天发表在《国家科学院学报》(PNAS)上。该研究中使用的软件,以及超过1000个冠状病毒基因组和计数的分类,在www.fluxus-technology.com免费提供。
Variant ‘A’, most closely related to the virus found in both bats and
pangolins, is described as “the root of the outbreak” by researchers.
Type ‘B’ is derived from ‘A’, separated by two mutations, then ‘C’ is in
turn a “daughter” of ‘B’.
研究人员说,"B"变种对东亚的本地化可能是由"创始人效应"引起的:当病毒从一小群孤立的感染中建立一种新类型时,就会发生一种基因瓶颈。
Researchers say the localisation of the ‘B’ variant to East Asia
could result from a “founder effect”: a genetic bottleneck that occurs
when, in the case of a virus, a new type is established from a small,
isolated group of infections.
福斯特认为,还有另一种解释值得考虑。"武汉B型病毒在免疫学或环境上可以适应东亚大部分人口。它可能需要变异,以克服东亚以外的阻力。在这个初始阶段,我们看到东亚的突变率比其他地方要慢。
Forster argues that there is another explanation worth considering.
“The Wuhan B-type virus could be immunologically or environmentally
adapted to a large section of the East Asian population. It may need to
mutate to overcome resistance outside East Asia. We seem to see a slower
mutation rate in East Asia than elsewhere, in this initial phase.”
他补充说:"我们详述的病毒网络是流行病早期阶段的快照,在COVID-19的进化路径被大量突变所掩盖之前。这就像在行动中捕捉一个初的超新星。
He added: “The viral network we have detailed is a snapshot of the
early stages of an epidemic, before the evolutionary paths of COVID-19
become obscured by vast numbers of mutations. It’s like catching an
incipient supernova in the act.”
自今天进行PNAS研究以来,研究小组将其分析扩展到1,001个病毒基因组。福斯特说,虽然还有待同行审查,但最新研究表明,COVID-19的首次感染和传播发生在9月中旬至12月初之间。
Since today’s PNAS study was conducted, the research team has
extended its analysis to 1,001 viral genomes. While yet to be
peer-reviewed, Forster says the latest work suggests that the first
infection and spread among humans of COVID-19 occurred between
mid-September and early December.
研究人员使用的植物遗传网络方法——允许在一个简单的图形中同时可视化数百种进化树——于1979年在新西兰率先出现,然后由德国数学家在20世纪90年代开发。
The phylogenetic network methods used by researchers – allowing the
visualisation of hundreds of evolutionary trees simultaneously in one
simple graph – were pioneered in New Zealand in 1979, then developed by
German mathematicians in the 1990s.
1998年,考古学家科林·伦弗鲁教授注意到了这些技术,他是PNAS新研究的合著者。伦弗鲁接着在剑桥大学建立了世界上第一个古遗传学研究小组。
These techniques came to the attention of archaeologist Professor
Colin Renfrew, a co-author of the new PNAS study, in 1998. Renfrew went
on to establish one of the first archaeogenetics research groups in the
world at the University of Cambridge.
《COVID-19: genetic network analysis provides ‘snapshot’ of pandemic origins》
Study charts the “incipient supernova” of COVID-19 through genetic mutations as it spread from China and Asia to Australia, Europe and North America. Researchers say their methods could be used to help identify undocumented infection sources.
Researchers from Cambridge, UK, and Germany have reconstructed the
early “evolutionary paths” of COVID-19 in humans – as infection spread
from Wuhan out to Europe and North America – using genetic network
techniques.
By analysing the first 160 complete virus genomes to be sequenced
from human patients, the scientists have mapped some of the original
spread of the new coronavirus through its mutations, which creates
different viral lineages.
“There are too many rapid mutations to neatly trace a COVID-19 family
tree. We used a mathematical network algorithm to visualise all the
plausible trees simultaneously,” said geneticist Dr Peter Forster, lead
author from the University of Cambridge.
“These techniques are mostly known for mapping the movements of
prehistoric human populations through DNA. We think this is one of the
first times they have been used to trace the infection routes of a
coronavirus like COVID-19.”
The team used data from virus genomes sampled from across the world
between 24 December 2019 and 4 March 2020. The research revealed three
distinct “variants” of COVID-19, consisting of clusters of closely
related lineages, which they label ‘A’, ‘B’ and ‘C’.
Forster and colleagues found that the closest type of COVID-19 to the
one discovered in bats – type ‘A’, the “original human virus genome” –
was present in Wuhan, but surprisingly was not the city’s predominant
virus type.
Mutated versions of ‘A’ were seen in Americans reported to have lived
in Wuhan, and a large number of A-type viruses were found in patients
from the US and Australia.
Wuhan’s major virus type, ‘B’, was prevalent in patients from across
East Asia. However, the variant didn’t travel much beyond the region
without further mutations – implying a "founder event" in Wuhan, or
“resistance” against this type of COVID-19 outside East Asia, say
researchers.
The ‘C’ variant is the major European type, found in early patients
from France, Italy, Sweden and England. It is absent from the study’s
Chinese mainland sample, but seen in Singapore, Hong Kong and South
Korea.
The new analysis also suggests that one of the earliest introductions
of the virus into Italy came via the first documented German infection
on January 27, and that another early Italian infection route was
related to a “Singapore cluster”.
Importantly, the researchers say that their genetic networking
techniques accurately traced established infection routes: the mutations
and viral lineages joined the dots between known cases.
As such, the scientists argue that these “phylogenetic” methods could
be applied to the very latest coronavirus genome sequencing to help
predict future global hot spots of disease transmission and surge.
“Phylogenetic
network analysis has the potential to help identify undocumented
COVID-19 infection sources, which can then be quarantined to contain
further spread of the disease worldwide,” said Forster, a fellow of the
McDonald Institute of Archaeological Research at Cambridge, as well as
the University’s Institute of Continuing Education.
The findings are published today in the journal Proceedings of the National Academy of Sciences (PNAS).
The software used in the study, as well as classifications for over
1,000 coronavirus genomes and counting, is available free at www.fluxus-technology.com.
Variant ‘A’, most closely related to the virus found in both bats and
pangolins, is described as “the root of the outbreak” by researchers.
Type ‘B’ is derived from ‘A’, separated by two mutations, then ‘C’ is in
turn a “daughter” of ‘B’.
Researchers say the localisation of the ‘B’ variant to East Asia
could result from a “founder effect”: a genetic bottleneck that occurs
when, in the case of a virus, a new type is established from a small,
isolated group of infections.
Forster argues that there is another explanation worth considering.
“The Wuhan B-type virus could be immunologically or environmentally
adapted to a large section of the East Asian population. It may need to
mutate to overcome resistance outside East Asia. We seem to see a slower
mutation rate in East Asia than elsewhere, in this initial phase.”
He added: “The viral network we have detailed is a snapshot of the
early stages of an epidemic, before the evolutionary paths of COVID-19
become obscured by vast numbers of mutations. It’s like catching an
incipient supernova in the act.”
Since today’s PNAS study was conducted, the research team has
extended its analysis to 1,001 viral genomes. While yet to be
peer-reviewed, Forster says the latest work suggests that the first
infection and spread among humans of COVID-19 occurred between
mid-September and early December.
The phylogenetic network methods used by researchers – allowing the
visualisation of hundreds of evolutionary trees simultaneously in one
simple graph – were pioneered in New Zealand in 1979, then developed by
German mathematicians in the 1990s.
These techniques came to the attention of archaeologist Professor
Colin Renfrew, a co-author of the new PNAS study, in 1998. Renfrew went
on to establish one of the first archaeogenetics research groups in the
world at the University of Cambridge.